![]()
![]()
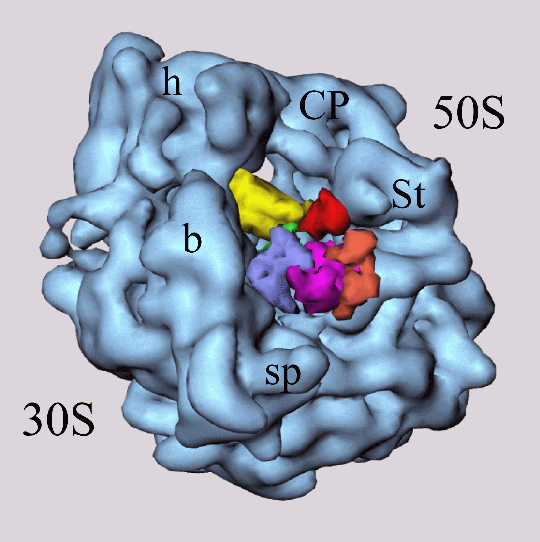
Fig. 1 (click to enlarge): EF-G bound to the ribosome, visualized by
difference mapping of 3D cryo-EM image reconstructions at 17 A
resolution. The Figure was created by Rajendra K. Agrawal in Joachim
Frank's lab. The colors code for the individual domains of EF-G (see
Fig. 2a below). Individual sites on the 30S and 50S ribosomal subunit
are also marked.
Frank's work has recently produced visually most intriguing evidence for induced-fit conformational differences in the form of EM image reconstructions of bound EF-G. These images, which were obtained from difference maps of free ribosomes and of ribosome-(fusidic acid)-EF-G complexes, are clearly incompatible with the shape of the crystal structure (see Fig. 2a). As a step towards a detailed understanding of the interactions of EF-G with the ribosome, and to identify the changes induced by binding, we refined an atomic resolution model of the bound protein against cryo EM data at 17 A resolution on the basis of information from the free structure of EF-G, secondary structure prediction methods, and a comparison with the structure of EF-Tu (Fig. 2).
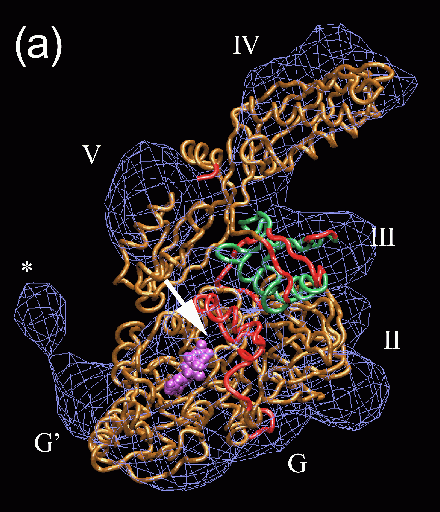


Fig. 2 (click to enlarge): Rigid-body and flexible docking of EF-G. (a)
The completed structure of EF-G (tube representation), fitted into the
EM density map with the Situs package.
The colors code for the original crystal coordinates (brown) of EF-G in
complex with GDP, coordinates of domain III (green) extracted from the
nucleotide-free structure, and coordinates added by secondary structure
prediction (red). The position of EF-G's structural domains is
indicated. The arrow points to the gap between Thr64 in the switch 1
loop (red) and the nearby beta phosphate of GDP. The EM density (blue)
in Fig. 2 is contoured at 14% of the maximum value. The asterisk
denotes a region of positive density that can be attributed to a
conformational change in the ribosome. EF-G is rotated to visualize the
GDP nucleotide (purple). (b) The flexibly fitted model of EF-G using
five pairs of point landmarks (grey spheres). The arrows point to
notable structural differences. (c) The final optimized protein
structure (brown tube representation). Also represented by spheres are
the positions of ten point landmarks (color and size-coded to
distinguish their resolution and weight, see the literature below).
![]()