![]()
![]()
![]()
This software is copyrighted, (c) 1995-1997 by Willy Wriggers under the terms of the legal statement in the distribution.
![]()
![]()
Hingefind is an algorithm for the identification of domain movements and their characterization and visualization by effective rotation axes (hinges). The program compares two known structures (e.g. two different crystal structures of a protein or the results of molecular dynamics simulations) and partitions the protein with a prespecified tolerance in preserved subdomains. It then determines effective rotation axes which characterize the domain movements with respect to the reference domain ("rigid core"). With little modification, the parts of the algorithm can also be used alone, i.e. one can assign domains manually and let the algorithm determine the movements between the domains, or one can use the algorithm to partition a protein into preserved subdomains. The method does not require any previous knowledge about functionally relevant domains or hinged domain motions, however a critical analysis of the results using the program output is recommended. The output files provide information about the accuracy of the found effective rotation. The variety of options and tolerances allow to find an optimal partitioning. The user can assess the validity of the depicted movement and change the script if necessary.
![]()
Currently, users have the choice of two implementations of Hingefind. The implementations are described at the following web sites:
![]()
 (click to enlarge image)
(click to enlarge image)
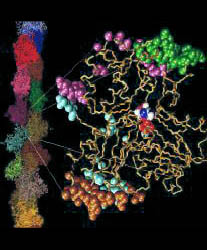
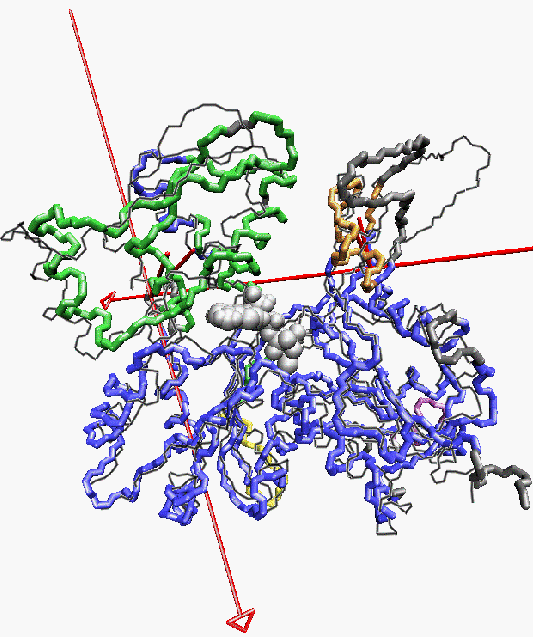
Backbone trace of Lorenz et al. F-actin structure ([JMB 1993 234:826], colored) compared to Kabsch et al. crystal structure ([Nature 1990 347:37], black line). The nucleotide and divalent cation of the comparison structure are rendered as grey van-der-Waals spheres. Five segments have been determined by "slo" mode partitioning at 3.76 A resolution (90% of initial rms-deviation). The color of the tubes codes for the partitioned segments found: reference-segment 1 (blue), segment 2 (green), segment 3 (orange), segment 4 (yellow), segment 5 (purple), no segment assigned (grey). The two structures are superimposed by a least-squares fit of segment 1. For segment 2 and segment 3, the rotation axis and "pivot" point-COM connecting lines of movements relative to segment 1 are shown as red tubes. The arrow indicates a right-handed rotation which transforms the COM of the respective segment of F-actin on the COM of the segment in G-actin. The rotation angle of segment 2 is 11.5 degrees. The rotation angle of segment 3 is 12.9 degrees. The domain movements yield a closure of the nucleotide binding cleft in the Lorenz structure. Segments 4 and 5 comprise only few residues.
![]()
The algorithm is separated in two parts: the "partitioning" and the "rotational fit" section. The "partitioning" part determines domains with preserved structure in the two compared coordinate sets, depending on a prespecified tolerance. The method uses the least-squares fit method (W. Kabsch, 1976) to superimpose structures. A domain is found in a iterative "adaptive selection" procedure, in which poor matching residues are excluded from the domain and good matches are included (in "slo" mode partitiononing, available in the X-PLOR version, only the heaviest connected set is considered, maintaining the connectivity of the changing domain). After a partitioning of the protein the rigid domains are sorted by their size and domains with a size larger than a cutoff considered for further evaluation.
The "rotational fit" method attempts to locate a rotation axis which characterize the movement of the domain as a hinged rigid body motion. The problem is to find an approximation of the rigid body motion with the constraint that transformations are not allowed, only rotations about the unknown hinge axis. Note that the the rotation about an axis without translation, in general, will not yield the closest fit of the Kabsch least-squares method. The accuracy of the approximation can be assessed by comparing the least-squares fit with the proposed rotational fit. The relative error (in percent) is computed as
where v is the COM separation of the domain in both positions.
The constructed rotation upholds (similar to a least-squares fit) the removal of the COM difference between the two coordinate sets, but approximates the rotation relative to a least-squares fit. Thus, the validity of the approximation can also be assessed by checking the "projection angle" beta between the least-squares rotation axis and the hinge axis, which should be small. One finds that the method works best for larger domains comprising several secondary structure elements. The projection angle beta and the relative error can be found in the output of the Hingefind runs.
For more information see also the domain movements site.
![]()
This software is not supported. Comments regarding the algorithm should be addressed to the author: wriggers >>at<< biomachina.org.
![]()